

Learning Objective
This module outlines the legislative and regulatory context for the development of generic drugs and describes the essential role of the Abbreviated New Drug Application (ANDA) in gaining marketing approval. The use of information in the ‘Orange Book’ is explained, as is the role of patent certification in the application. The importance of establishing bioequivalence between a generic and its reference product is emphasised. The module specifies the content and format requirements for an ANDA submission and describes the FDA’s review and approval process. An outline is given of the Generic Drug User Fee Amendments (GDUFA) and the law’s effects on industry players.
The module is up to date with the many final and draft publications, recently released by the FDA, that provide guidance for industry on applications for approval of generic drugs. It is also up to date with the provisions of the second authorisation of GDUFA, applicable in fiscal years 2018 to 2022.
Who will benefit from this module?
This module will benefit staff working in regulatory affairs, medical affairs, clinical development, quality/CMC, analytical methods, and quality assurance departments, and other personnel who contribute to the development and registration of generic drugs.
Learning Objectives
- List the criteria for therapeutic equivalence of drugs
- Outline the types of patent classification for an ANDA submission
- Explain how to use the Orange Book in the development of a generic drug
- Describe methods for determining bioequivalence of drug products
- Outline the content and format requirements for an ANDA submission
- Describe the ANDA review and approval process
- Outline the provisions of the Generic Drug User Fee Amendments and summarise their effects on generics sponsors
Module Outline
Module overview - An outline of the module’s scope and objectives, and notes on terminology.
Generic Drugs and the ANDA - An overview of the legislative and regulatory context for the development and approval of generic drugs, particularly the Hatch-Waxman Act; a summary of the criteria for therapeutic equivalence of drugs; obtaining guidance from the FDA; controlled correspondence.
Patent certification - The role of patent certification in an ANDA submission, the different types of certification, what happens when a patent is challenged, and the circumstances under which marketing exclusivity may be afforded to a generics sponsor.
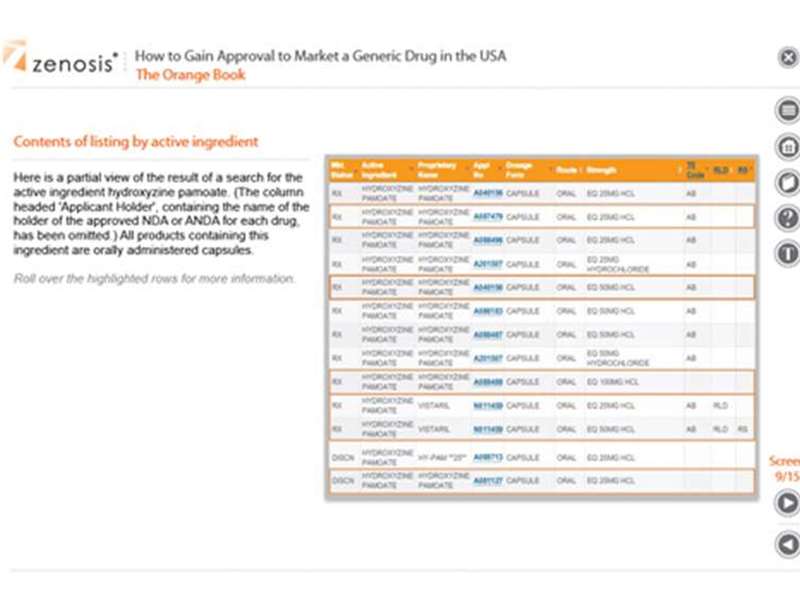
The Orange Book - The use of the Orange Book in generic drug development, the format and content of the Book’s listings, and how to extract information for an ANDA.
Bioequivalence - The crucial importance of establishing bioequivalence with a reference listed drug; tests of bioavailability and bioequivalence; the statistical criteria for bioequivalence; waivers of in-vivo studies.
ANDA compilation and submission - Planning and managing an ANDA project; regulatory requirements on content and format; quality (CMC), labeling, and bioequivalence information; submitting an ANDA to the FDA’s Office of Generic Drugs.
ANDA review and approval - The process of review by the FDA; review duration and success rate; communication between applicant and FDA; expedited review; petitions; amendments and easily correctable deficiencies; outcomes of review, and the applicant’s options in response to those outcomes.
The Generic Drugs User Fee Amendments - The types of fees that the generics industry must now pay to the FDA; requirements for self-identification of generics industry players; the FDA’s performance goals for review and inspection; changes brought about by GDUFA II.
Assessment - Multiple-choice mastery assessment.